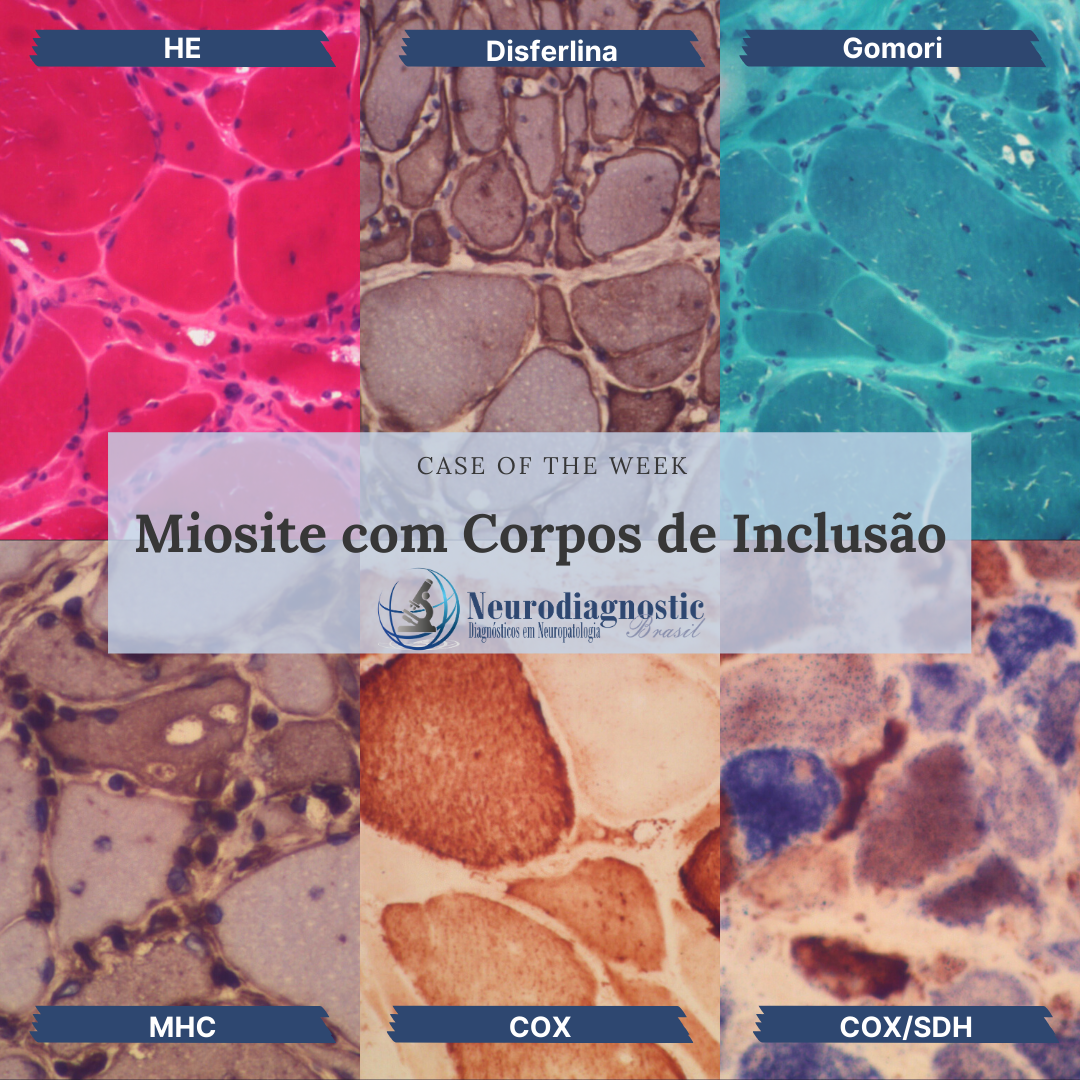
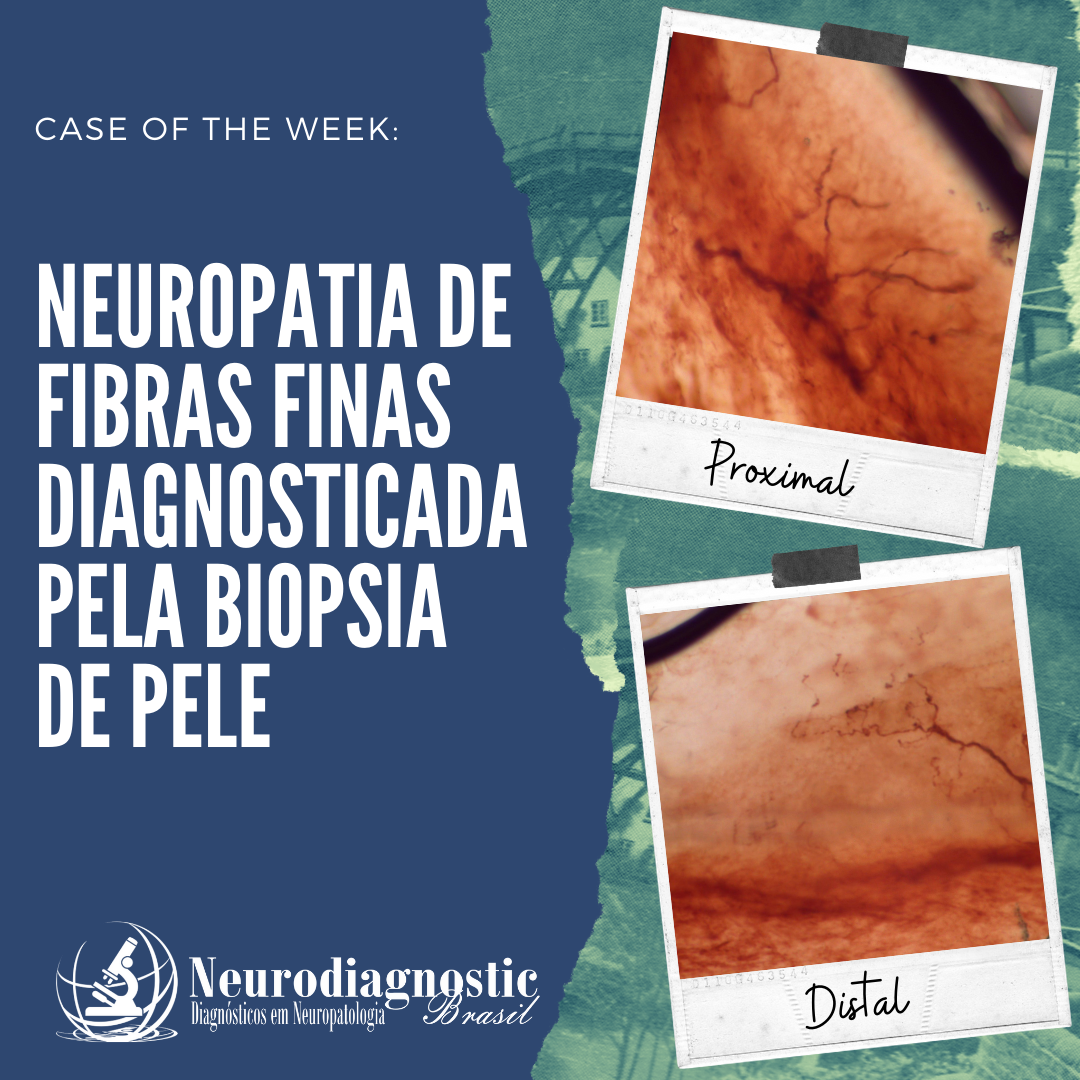
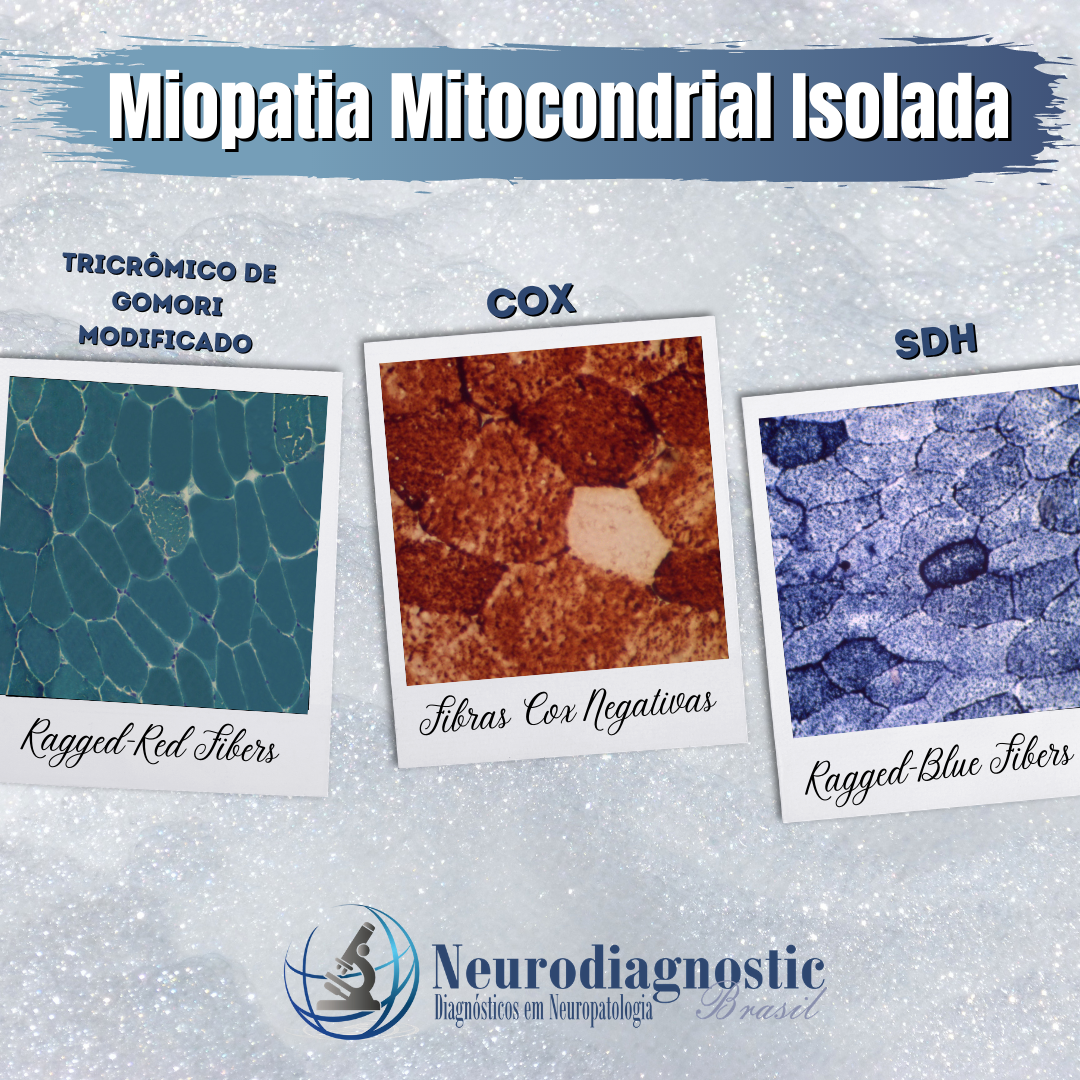
O sistema nervoso (SN) humano é de longe o sistema mais complexo do corpo humano, sendo formado por uma rede de mais de 100 milhões de células nervosas (neurônios), auxiliadas por muito mais células gliais. Cada neurônio possui, em média, 1000 interconexões com outros neurônios, formando um sistema de comunicação bastante complexo. Os neurônios são agrupados em circuitos. Como os circuitos eletrônicos, os circuitos neurais são combinações altamente específicas de elementos que constituem sistemas de vários tamanhos e complexidades. Embora um circuito neural possa ser único, na maioria dos casos é uma combinação de dois ou mais circuitos que interagem para gerar uma função. Uma função neural é um conjunto de processos coordenados destinados a produzir um resultado definido. Vários circuitos elementares podem ser combinados para formar sistemas de ordem superior.
Estruturalmente, o tecido nervoso consiste em dois tipos de células: células nervosas, ou neurônios, que geralmente mostram vários processos longos, e vários tipos de células gliais, que têm processos curtos, suportam e protegem os neurônios e participam de atividade neural, nutrição neural e de processos de defesa do sistema nervoso central. O estudo do tecido nervoso recentemente progrediu rapidamente, devido ao uso de marcadores que identificam neurônios e células da glia. Os neurônios respondem às mudanças ambientais (estímulos) alterando os potenciais elétricos que existem entre as superfícies interna e externa de suas membranas. As células com essa propriedade (por exemplo, neurônios, células musculares, algumas células glandulares) são chamadas de excitáveis ou irritáveis. Os neurônios reagem prontamente aos estímulos com uma modificação do potencial elétrico que pode ser restrito ao local que recebeu o estímulo ou pode ser espalhado (propagado) por todo o neurônio pela membrana plasmática. Essa propagação, chamada de potencial de ação ou impulso nervoso, é capaz de viajar longas distâncias; ele transmite informações a outros neurônios, músculos e glândulas. Ao criar, analisar, identificar e integrar informações, o sistema nervoso gera duas grandes classes de funções: estabilização das condições intrínsecas (por exemplo, pressão arterial, conteúdo de O2 e CO2, pH, níveis de glicose no sangue e níveis hormonais) do organismo dentro de faixas normais e padrões de comportamento (por exemplo, alimentação, reprodução, defesa, interação com outras criaturas vivas).
Fonte: Mesher AL. Basic Histology Text and Atlas.2015