A maioria dos tumores hipofisários são pouco agressivos, mas podem causar diversos transtornos ao alterar a produção de hormônio de inúmeras glândulas em nosso corpo ou ainda alguns sintomas neurológicos como: visão dupla e perda da acuidade, perda de visão periférica, cegueira repentina, dor de cabeça, dor ou dormência no rosto, tontura, desmaios, etc.
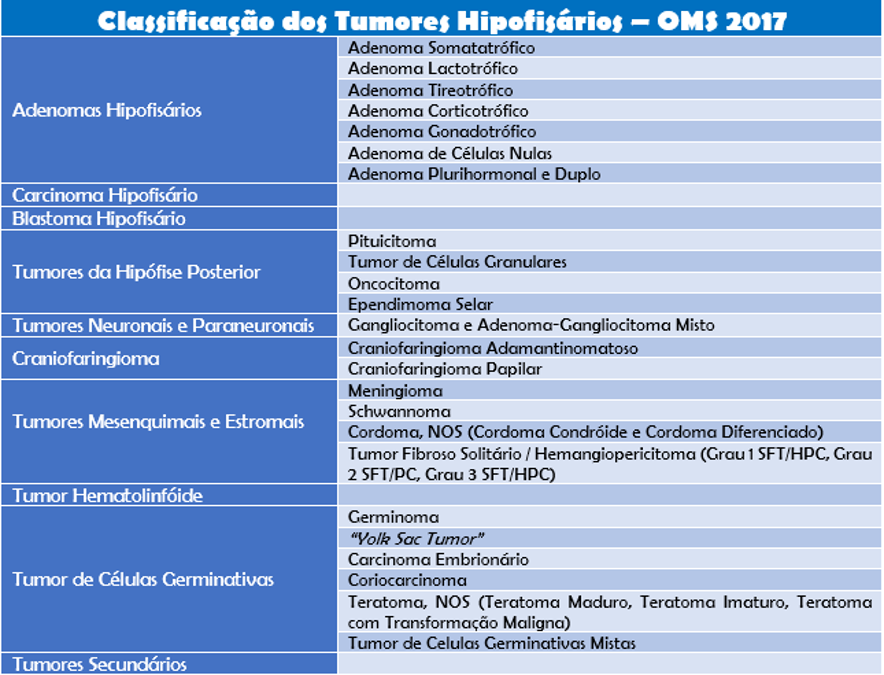
A Organização Mundial da Saúde (OMS) liberou em 2017 uma nova classificação de tumores hipofisários. Esta nova classificação oferece informações de grande relevância ao tratamento e ao prognóstico do paciente.
A nova classificação é feita preferencialmente de acordo com a linhagem celular ao invés da produção hormonal realizada pela hipófise. Outro ponto importante enfatizado pela nova classificação é a definição da imunohistoquímica em combinação com outras colorações especiais como as principais técnicas necessárias à sua classificação.
Esta nova classificação ficou mais prática, fornecendo informações definitivas ao diagnóstico e valiosos dados prognósticos que auxiliam muito na orientação do tratamento a ser definido pela equipe médica. Lesões mais agressivas devem ser identificadas com base na histopatologia, índice mitótico, índice de marcação do anticorpo Ki-67, bem como a infiltração do tumor nas demais áreas cerebrais.
FONTE: LOPES, M. Beatriz S. The 2017 World Health Organization classifiction os tumors of the pituitary gland: a summary. Acta Neuropathol., n. 134, p. 521-535. Springer-Verlag GmbH Germany 2017.
Em muitos tumores do Sistema Nervoso Central, a localização anatômica, os detalhes microscópicos e outros aspectos de sua apresentação clínica possuem forte influência sobre a probabilidade relativa de seus respectivos diagnósticos. Mesmo com tais considerações, os patologistas frequentemente são confrontados com neoplasias que apresentam caracteres morfológicos que restringem o diagnóstico definitivo.
O estudo imunohistoquímico é uma ferramenta complementar que fornece subsídios muito significativos na caracterização de entidades e/ou doenças distintas. As informações adquiridas neste tipo de estudo permitem complementar o que as variáveis morfológicas não permitiram definir com clareza. Assim é possível reconhecer antígenos de superfície, intracitoplasmáticos ou nucleares que são peculiares para cada uma das entidades e alcançar uma conclusão definitiva sobre o diagnóstico.
Este é um método avançado baseado em uma reação antígeno-anticorpo, que produz uma coloração específica quando o anticorpo realiza ligação com o antígeno pesquisado. Pode ser efetuado com material fixado em formol e emblocado em parafina, fornecendo uma grande vantagem quando comparado a outros métodos.
Uma abordagem integrada de estudos imunohistoquímicos e colorações especiais em tumores pouco diferenciados e morfologicamente ambíguos é extremamente relevante para a definição diagnóstica do tumor. Além da caracterização de tumores pouco diferenciados, os estudos imunohistoquímicos também permitem classificar com clareza o grau de agressividade e velocidade de crescimento do tumor em questão, bem como detectar com alto grau de precisão o produto de mutações genéticas importantes para orientação terapêutica, como a mutação TP53 e o IDH, de grande relevância no estudo de gliomas e também eventuais sítios primários em caso de tumores metastáticos.
As análises imunohistoquímicas devem ser adaptadas de acordo com informações como a localização anatômica, características morfológicas e outros aspectos clínicos observados previamente. A correlação com o quadro clínico do paciente pode trazer informações de grande relevância ao diagnóstico e aumentar as chances de sucesso dos tratamentos indicados. Ainda assim, estes estudos são totalmente dependentes da interpretação do patologista, exigindo uma grande base de conhecimento de análise histopatológica.
Portanto, a fim de garantir a definição diagnóstica e aumentar a chance de sucesso dos tratamentos indicados a cada paciente, a Organização Mundial da Saúde (OMS) orienta a realização destes estudos para as análises anatomopatológicas. Mesmo com o avanço das técnicas dos estudos moleculares, estes não substituem as análises imunohistoquímicas. Os diferentes métodos diagnósticos devem ser usados seletivamente, a critério do patologista, para fornecer soluções efetivas e abrangentes à casos específicos.
FONTE: WICK, MR. Immunohistochemical approaches to the diagnosis of undifferentiated malignnt tumors. Annals of Diagnostic Pathology, n. 12, p 72-84, 2008.
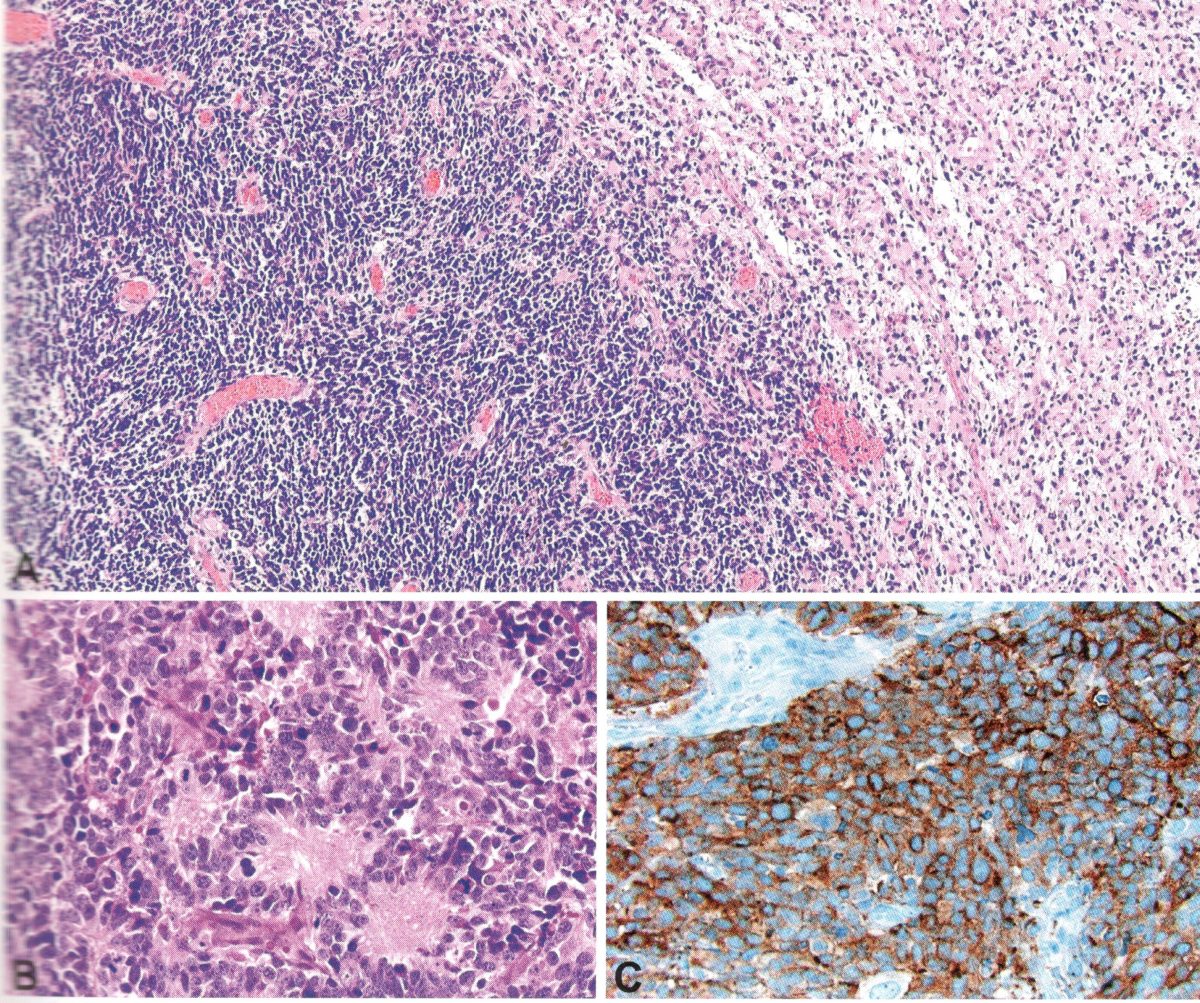
Os Astrocitomas Pilocíticos constituem o grupo de gliomas mais frequente em crianças e adolescentes. Estas neoplasias não apresentam predileção significativa pelo sexo, mas alguns estudos descrevem incidência ligeiramente superior em pacientes do sexo masculino.
Este tipo de neoplasia frequentemente está localizada nas estruturas da linha média (vias oculares, hipotálamo e tronco cerebral) e na fossa posterior, mas também podem ser encontrados em outras estruturas ao longo do neuroeixo. São normalmente bem delimitados e apresentam baixa atividade proliferativa, frequentemente apresentando um componente cístico.
Os Astrocitomas Pilocíticos são tipicamente classificados como um tumor OMS Grau I e seu tratamento pode ser exclusivamente cirúrgico desde que sua localização permita tal intervenção sem riscos significativos ao paciente.
Apesar de ser encontrado predominantemente em crianças, é possível que o Astrocitoma Pilocítico seja encontrado em adultos jovens, mas raramente atingem indivíduos com mais de 50 anos.
Astrocitomas pilocíticos podem produzir déficits neurológicos focais ou sinais não localizatórios, como macrocefalia, dores de cabeça, endocrinopatia e aumento da pressão intracraniana devido ao efeito de massa local ou obstrução do sistema ventricular. Crises epilépticas podem ocorrer, mas são incomuns, já que as lesões dificilmente envolvem o córtex cerebral. Perda visual é um sintoma frequente, visto que as lesões podem acometer as vias visuais.
Inicialmente, apenas os aspectos morfoestruturais eram considerados para o diagnóstico de gliomas. Atualmente a caracterização molecular das lesões têm oferecido subsídios relevantes para orientação da conduta e comportamento biológico.
Entre os aspectos moleculares descritos entre os pacientes com astrocitoma pilocítico estão as alterações genéticas listadas abaixo:
GENE
Alteração
% dos Tumores
Utilidade Diagnóstica
BRAF e KIAA1549
Duplicações sequenciais que resultam em proteínas de fusão KIAA1549-BRAS, todas contendo o domínio da BRAF quinase e com a regulação do domínio regulatório do BRAF N-Terminal
>70%
Comum em astrocitomas pilocíticos, particularmente os cerebelares; raro em outras formas de tumor.
BRAF e outros genes diversos
Deleções e translocações resultando em proteínas de fusão BRAF quinase, todas possuindo o domínio BRAF quinase e com o domínio BRAF regulatório substituído pela parte N-Terminal de outro gene
~5%
Ocorre em astrocitomas pilocíticos; extremamente raro em outras entidades.
BRAF
Mutação V600E
~5%
Ocorre principalmente em astrocitomas pilocíticos supratentoriais; também em gangliogliomas, xantoastrocitomas pleomórficos.
NF1
Perda do tipo selvagem. Mutação hereditária acumulada.
~8%
Tipicamente células germinativas; muito associado ao astrocitoma pilocítico do nervo ótico.
FGFR1
Mutação
<5
Encontrado principalmente em astrocitomas pilocíticos da linha média; frequência não estabelecida em outras entidades.
FGFR1
Fusões / duplicações sequenciais internas
<5
Raro no astrocitoma pilocítico; também observado em outros gliomas de baixo grau.
Família NTRK
Fusões
~2%
Raro no astrocitoma pilocítico; frequência não estabelecida em outras entidades.
KRAS
Mutação
Casos Únicos
Raro no astrocitoma pilocítico; frequência não estabelecida em outras entidades.
RAF1
Fusões com consequências similares às fusões BRAF, ou seja, perda do domínio regulatório e substituição pelo final do N-Terminal do SGRAP3
Casos Únicos
Raro no astrocitoma pilocítico; frequência não estabelecida em outras entidades.
Tabela 1 – Alterações genéticas que afetam a via do MAPK em astrocitomas pilocíticos e sua utilidade diagnóstica. (adaptado de WHO, 2016).
Fonte: WHO classification of tumors of the central nervous system / edited by David N. Louis, Hiroko Ohgaki, Otmar D. Wiestler, Webster K. Cavanee – Revised 4th Edition. Lyon, 2016.
Se você ou algum conhecido possui dores de cabeça persistentes associadas a náuseas e vômitos, notou alguma perda de força ou rigidez muscular, teve alterações visuais, auditivas ou de equilíbrio, passou a ter dificuldades de fala ou compreensão, tem notado movimentos involuntários ou perda de coordenação, fique atento e procure um médico de confiança. Esses são sintomas comuns de algumas doenças cerebrais.
Se for identificada a presença de um tumor cerebral, confie em seu médico e siga as orientações clínicas. A medicina evolui constantemente e de forma extremamente rápida. Nestes casos, é necessário agilidade. É preciso identificar o tipo do tumor para que o tratamento seja efetivo. O diagnóstico preciso do tipo de tumor que o paciente apresenta, orienta os médicos na adoção das melhores práticas existentes na atualidade e melhora significativamente o prognóstico (chances de cura) do paciente.
O primeiro passo após a detecção de uma lesão suspeita de tumor cerebral, é procurar um neurocirurgião. Ele vai definir a melhor estratégia para abordar a lesão e encaminhá-la para análise, a fim de definir o diagnóstico e a conduta. A estratégia cirúrgica a ser adotada vai depender do tamanho e localização da lesão, bem como das condições clínicas do paciente. Assim é possível ter clareza na conduta e nos tratamentos a serem adotados dali em diante.
Em linhas gerais, há duas formas de abordar a lesão: realizando uma ressecção ou uma biopsia, que pode ser aberta ou estereotáxica.
A ressecção é a retirada integral (ou da maior parte possível) do tumor. Apesar de retirar a maior parte da lesão de forma cirúrgica, esta abordagem depende da localização do tumor e/ou da condição clínica do paciente. Nem sempre é possível realizar este tipo de abordagem, devido aos riscos envolvidos neste processo e/ou à dificuldade de acesso à lesão.
Em caso de impossibilidade de ressecção total (ou parcial) da lesão, ou quando a lesão ainda está em fase inicial de investigação, os médicos programam a retirada de uma pequena amostra da lesão apenas para fins diagnósticos. Nesses casos, o paciente é submetido à uma biopsia (estereotáxica ou aberta). Este processo é menos invasivo e traz menos riscos aos pacientes. O fragmento retirado é, na maioria das vezes, suficiente para análise da lesão e orientação do tratamento.
A peça retirada em cirurgia deve ser enviada ao neuropatologista para definição diagnóstica do tipo de lesão e posterior orientação de conduta a ser seguida. Através de técnicas altamente avançadas, o material é processado e passa por análises morfológica, histoquímica e imuno-histoquímica. Se necessário, também pode ser realizado estudo molecular para caracterização. A partir de todos esses estudos, o neuropatologista consegue definir com clareza o tipo de tumor e o paciente pode dar sequência ao seu tratamento.
É a partir do laudo neuropatológico que os oncologistas e radioterapeutas conseguirão traçar as melhores estratégias de tratamento ao paciente. Por isso é essencial que este processo seja ágil e o paciente consiga iniciar o tratamento o quanto antes.
Informe-se sobre as melhores alternativas e confie na equipe que escolher para lhe auxiliar durante este processo. Eles vão guiá-lo pelos melhores caminhos a serem seguidos e estarão com você nos momentos em que mais precisar.
O Glioblastoma é um glioma de alto grau com predomínio de diferenciação astrocítica. Esse tumor apresenta atipias nucleares e, na maioria dos casos, pleomorfismo celular (variação no formato), atividade mitótica proeminente e um padrão de crescimento difuso, assim como proliferação microvascular/endotelial e/ou áreas necróticas.
Estes tumores se desenvolvem com grande rapidez e os sintomas ocasionados dependem da localização da lesão. Inicialmente manifesta déficits neurológicos focais, como hemiparesia (perda de força em um dos lados do corpo) ou afasia (alterações da fala) e aumento da pressão intra-craniana, entre outros. Mais da metade dos pacientes são diagnosticados com uma lesão cerebral após uma primeira crise epiléptica. Outros sintomas comuns são mudanças cognitivas e comportamentais, náuseas e vômitos, e cefaleias intensas e progressivas.
O Glioblastoma é a lesão cerebral maligna mais frequente em adultos. Ele representa aproximadamente 15% de todas as neoplasias intracranianas e entre 45% e 50% de todos os tumores cerebrais malignos primários.
Os Glioblastomas que não possuem mutação do gene IDH (IDH-Wildtype) são os mais comuns, representando aproximadamente 90% de todos os glioblastomas. Estes tumores podem se manifestar em pacientes de qualquer idade, mas afetam preferencialmente adultos entre 55 e 85 anos.
Mesmo com alto grau de mortalidade, alterações favoráveis em nível molecular como mutação do IDH e/ou Metilação do Promotor MGMT possuem frequentemente taxas de resposta mais positivas aos tratamentos existentes em relação às lesões que não possuem tais mutações.
NEURODIAGNOSTIC BRASIL
Fonte: WHO classification of tumors of the central nervous system / edited by David N. Louis, Hiroko Ohgaki, Otmar D. Wiestler, Webster K. Cavanee – Revised 4th Edition.
O Astrocitoma Difuso (OMS GRAU II) é composto por astrócitos bem diferenciados em uma matriz tumoral frequentemente microcística e desestruturada. Esta neoplasia apresenta crescimento lento e celularidade moderadamente elevada.
Na Classificação da OMS2016, a nomenclatura Astrocitoma de Baixo Grau foi desencorajada e a nomenclatura Astrocitoma Fibrilar também não foi mais recomendada, já que este é o tipo clássico de Astrocitoma Difuso e não está mais listado como uma variante.
Este tumor afeta de maneira mais comum adultos jovens e pode se localizar em qualquer região do Sistema Nervos Central, preferencialmente, no lobo frontal. Astrocitomas Difusos possuem a capacidade intrínseca de uma progressão maligna para um astrocitoma anaplásico e eventualmente um glioblastoma.
Apesar da análise histológica ser mais utilizada, estudos recentes sugerem que os parâmetros moleculares podem trazer informações de extrema importância para predizer o prognóstico da lesão.
Astrocitomas Difusos IDH-Mutantes representam aproximadamente 90% dos casos e antes da era molecular a média da sobrevida de um paciente com este diagnóstico foi descrita como sendo entre 6-8 anos. Porém, este cenário está sendo revisto no contexto do status da mutação do IDH.
Um estudo recente, que incluiu 683 casos diagnosticados como Astrocitomas Difusos IDH-Mutante, demonstrou uma média de sobrevida de 10,9 anos, demonstrando um curso bem mais favorável em relação às lesões que possuem IDH-Selvagem.
Imagem: WHO classification of tumors of the central nervous system / edited by David N. Louis, Hiroko Ohgaki, Otmar D. Wiestler, Webster K. Cavanee – Revised 4th Edition.
Sed diam nonummy nibh euismod tincidunt ut laoreet dolore magna aliquam erat volutpat. Quis nostrud exerci tation ulla. Ut wisi enim ad minim veniam. Eodem modo typi, qui nunc nobis videntur parum clari, fiant sollemnes in futurum. Lorem ipsum dolor sit amet, consectetuer adipiscing elit.