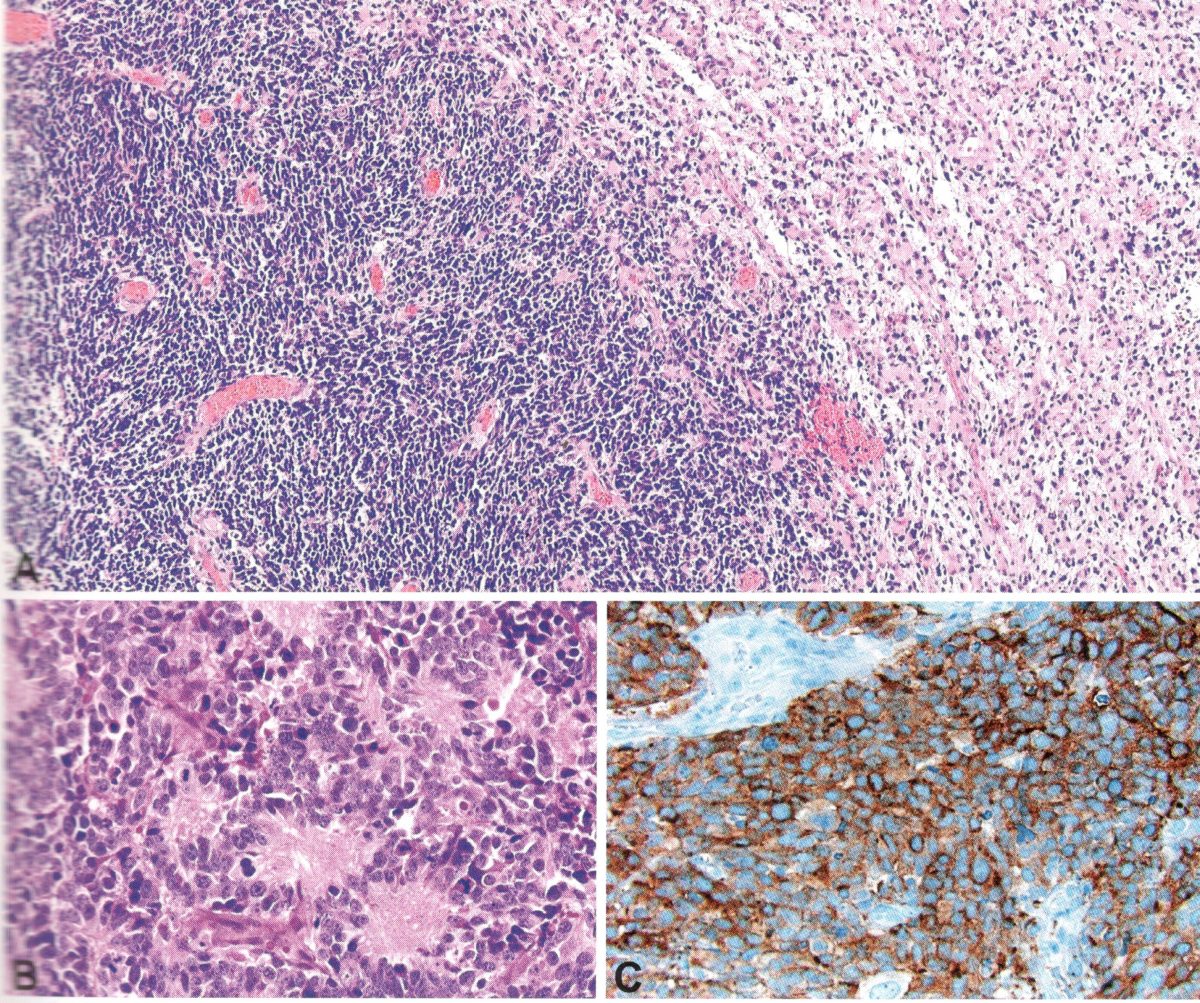
Os Astrocitomas Pilocíticos constituem o grupo de gliomas mais frequente em crianças e adolescentes. Estas neoplasias não apresentam predileção significativa pelo sexo, mas alguns estudos descrevem incidência ligeiramente superior em pacientes do sexo masculino.
Este tipo de neoplasia frequentemente está localizada nas estruturas da linha média (vias oculares, hipotálamo e tronco cerebral) e na fossa posterior, mas também podem ser encontrados em outras estruturas ao longo do neuroeixo. São normalmente bem delimitados e apresentam baixa atividade proliferativa, frequentemente apresentando um componente cístico.
Os Astrocitomas Pilocíticos são tipicamente classificados como um tumor OMS Grau I e seu tratamento pode ser exclusivamente cirúrgico desde que sua localização permita tal intervenção sem riscos significativos ao paciente.
Apesar de ser encontrado predominantemente em crianças, é possível que o Astrocitoma Pilocítico seja encontrado em adultos jovens, mas raramente atingem indivíduos com mais de 50 anos.
Astrocitomas pilocíticos podem produzir déficits neurológicos focais ou sinais não localizatórios, como macrocefalia, dores de cabeça, endocrinopatia e aumento da pressão intracraniana devido ao efeito de massa local ou obstrução do sistema ventricular. Crises epilépticas podem ocorrer, mas são incomuns, já que as lesões dificilmente envolvem o córtex cerebral. Perda visual é um sintoma frequente, visto que as lesões podem acometer as vias visuais.
Inicialmente, apenas os aspectos morfoestruturais eram considerados para o diagnóstico de gliomas. Atualmente a caracterização molecular das lesões têm oferecido subsídios relevantes para orientação da conduta e comportamento biológico.
Entre os aspectos moleculares descritos entre os pacientes com astrocitoma pilocítico estão as alterações genéticas listadas abaixo:
| GENE | Alteração | % dos Tumores | Utilidade Diagnóstica |
| BRAF e KIAA1549 | Duplicações sequenciais que resultam em proteínas de fusão KIAA1549-BRAS, todas contendo o domínio da BRAF quinase e com a regulação do domínio regulatório do BRAF N-Terminal | >70% | Comum em astrocitomas pilocíticos, particularmente os cerebelares; raro em outras formas de tumor. |
| BRAF e outros genes diversos | Deleções e translocações resultando em proteínas de fusão BRAF quinase, todas possuindo o domínio BRAF quinase e com o domínio BRAF regulatório substituído pela parte N-Terminal de outro gene | ~5% | Ocorre em astrocitomas pilocíticos; extremamente raro em outras entidades. |
| BRAF | Mutação V600E | ~5% | Ocorre principalmente em astrocitomas pilocíticos supratentoriais; também em gangliogliomas, xantoastrocitomas pleomórficos. |
| NF1 | Perda do tipo selvagem. Mutação hereditária acumulada. | ~8% | Tipicamente células germinativas; muito associado ao astrocitoma pilocítico do nervo ótico. |
| FGFR1 | Mutação | <5 | Encontrado principalmente em astrocitomas pilocíticos da linha média; frequência não estabelecida em outras entidades. |
| FGFR1 | Fusões / duplicações sequenciais internas | <5 | Raro no astrocitoma pilocítico; também observado em outros gliomas de baixo grau. |
| Família NTRK | Fusões | ~2% | Raro no astrocitoma pilocítico; frequência não estabelecida em outras entidades. |
| KRAS | Mutação | Casos Únicos | Raro no astrocitoma pilocítico; frequência não estabelecida em outras entidades. |
| RAF1 | Fusões com consequências similares às fusões BRAF, ou seja, perda do domínio regulatório e substituição pelo final do N-Terminal do SGRAP3 | Casos Únicos | Raro no astrocitoma pilocítico; frequência não estabelecida em outras entidades. |
Tabela 1 – Alterações genéticas que afetam a via do MAPK em astrocitomas pilocíticos e sua utilidade diagnóstica. (adaptado de WHO, 2016).
Fonte: WHO classification of tumors of the central nervous system / edited by David N. Louis, Hiroko Ohgaki, Otmar D. Wiestler, Webster K. Cavanee – Revised 4th Edition. Lyon, 2016.